Årsrapport til ALS Norge for 2020
Joel Glover
Institutt for medisinske basalfag, Universitetet i Oslo og Nasjonalt senter for stamcelleforskning, Oslo universitetssykehus
2020 var et utfordrende år for forskning ved Nasjonalt senter for stamcelleforskning. I mars ble det meste av aktivitet stengt ned pga koronavirus-pandemien, og restriksjoner som har vært på plass siden har hindret hos i å nå tilbake til full aktivitet med unntak av enkelte korte perioder.
Allikevel har vi klart å holde det gående og har gjort noen fremskritt takket være økonomisk støtte fra ALS Norge. Her gir vi en oppsummering av forskningsaktivitet vårt, støttet av ALS Norge.
1) Via Trygve Holmøy ved AHUS og Morten Andreas Horn ved OUS kontaktet vi 4 ALS pasienter med den bestemte ALS genmutasjon C9orf72, tok hudbiopsier, og igangsatt reprogrammering til induserte pluripotente stamceller (iPS celler). Reprogrammeringen vil være ferdig innen utgangen av januar 2021. I tillegg er det en 5. pasient med den samme mutasjon som det vil tas hudbiopsi av i februar 2021.
Ved dette kommer vi opp i totalt 6 norske ALS pasienter med en SOD-1 mutasjon og 6 norske ALS pasienter med en C9orf72 mutasjon som vi har laget iPS celler fra, med 4-5 iPS cellelinjer per pasient. Vi er derved blant et fåtall land som har laget såpass mange iPS cellelinjer fra ALS pasienter. Disse iPS celler er en uhyre viktig resurs for videre forskning på sykdomsmekanismer knyttet til de ulike ALS genmutasjonene, og vil også fremme forskningssamarbeid med utlandet.
2) Vi har jobbet videre med etablering av gode og standardiserte prosedyrer for differensiering av ulike typer humane celler fra iPS celler. Differensiering betyr at man tar iPS celler og styrer dem i laboratoriet til å utvikle seg til en bestemt celletype. Dette betyr i grunn at man etterligne det som skjer under fosterutviklingen, der stamceller gir opphav til alle kroppens celletyper, bare at man gjør det i en petriskål. Prosedyrene innebærer behandling av iPS cellene med ulike signalstoffer i ørsmå mengder og i nøye tidsbestemte rekkefølger, og er svært vanskelig å få til på en standardisert måte. Dette arbeidet krever mye innsats, men gevinsten er at man kan da skape de ulike celletyper som er involvert i ALS og kombinere dem in vitro (dvs i en petriskål eller annen type dyrkingskammer) på en måte som simulerer situasjonen i kroppen. Man kan derved lage det som kalles for en in vitro ALS modell, som gjør det mulig å studere sykdomsmekanismer på måter som ikke er mulig hos pasienter. De celletypene som vi nå har standardiserte differensieringsprosedyrer for er motoriske nerveceller, kortikale glutamaterge nerveceller, astrocytter, og mikroglia-celler. Motoriske nerveceller i hjernestammen og ryggmargen er nervecellene som dør pga ALS med lammelse som resultat, og kortikale glutamaterge nerveceller antas å være nervecelletypen som rammes av frontotemporal demens, som også kan oppstå sammen med ALS. Astrocytter er kjent for å ha en viktig signalfunksjon overfor motoriske nerveceller under ALS, og mikroglia-celler spiller også en viktig rolle når nerveceller begynner å gå til grunne. Disse celletyper representerer de viktigste som er involvert i ALS.
Vi jobber med differensiering av en celletype til, nemlig skjelettmuskelceller, som også er viktig ved ALS fordi motoriske nerveceller normalt er avhengig av kontakt med disse for å få nødvendig vekst- og overlevelsesfaktorer. Under ALS blir forskyningen av disse faktorene avbrutt når de motoriske nervecellene begynner å skades, fordi kontakten med muskelcellene svinner hen. Vi har ennå ikke lyktes med å få til en fullgod, standardisert differensieringsprosedyre for skjelettmuskelceller, men vi jobber med saken.
Vi har med andre ord etablert gode differensieringsprosedyrer for alle bortsett fra en celletype som er involvert i ALS, noe som de færreste forskningsgrupper i verden har klart.
3) Vi har jobbet videre med utvikling av vår in vitro ALS modell, der vi sår ut disse celletypene i en spesiallaget mikrofluidikk apparat, der vi kan ordne cellene i bestemte forhold til hverandre slik at vi kan simulere sykdomsprosesser som forekommer i kroppen og kan undersøke celletypene hver for seg under sykdomsutviklingen. Dette gir en stor gevinst når det gjelder å tolke hva som skjer, og vår modell er den mest utviklede i denne sammenheng som vi kjenner til internasjonalt. Den gjør det mulig blant annet å se spesifikt på hva som skjer ved kontaktene mellom motoriske nerveceller og muskelceller (der mange regner med at de første sykdomseffektene oppstår) og hva som skjer ved signalering mellom gliaceller og motoriske nerveceller (som er et viktig element i sykdomsutvikling hos mange former for ALS).
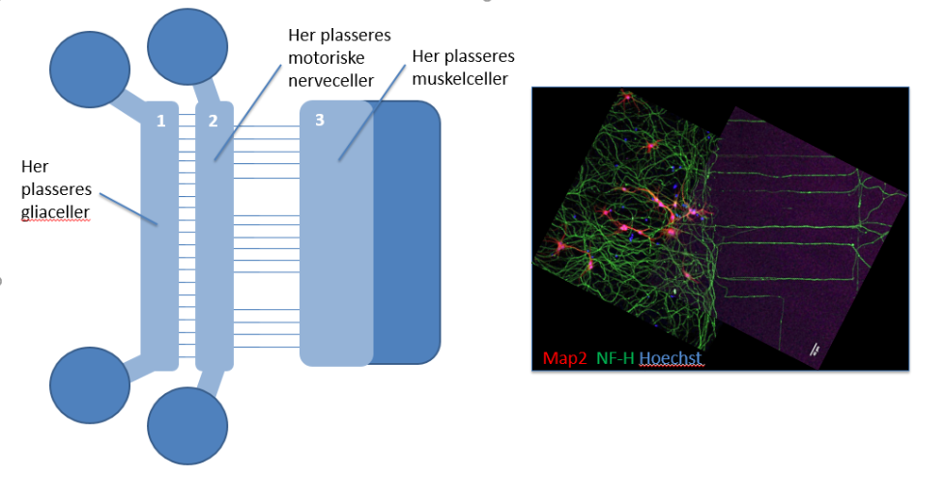
Figur 1. Vår mikrofluidikk-basert in vitro ALS modell. På venstre siden vises en tegning av mikrofluidikk-apparatet sett i fugleperspektiv. Det er 3 kamre der celler plasseres (1-3), farget med lyseblått (de mørke blå delene er portaler der man kan fôre cellene med dyrkningsmedium). Mellom kamrene finnes det kanaler (horisontale linjer) som gir mulighet for signaler å passere fra astrocytter til motoriske nerveceller (fra kammer 1 til kammer 2) og nervefibre å vokse fra motoriske nerveceller til muskelceller (fra kammer 2 til kammer 3). PÅ høyre siden er et bilde som viser motoriske nerveceller (farget i rosa) i kammer 2 og deres nervefibre (farget i grønt) som vokser gjennom kanalene til kammer 3, der de lager kontakter med muskelceller.
Når vi skal lage en in vitro FTD modell, sår vi ut kortikale glutamaterge nerveceller i både kammer 2 og 3. Kanalene mellom disse tillater nervefibervekst kun i en retning (fra kammer 2 til kammer 3), slik at vi kan kontroller hvilke nerveceller sender signaler til andre nerveceller.
4) I tillegg til å kunne bruke dette mikrofluidikk apparatet til å simulere hva som skjer når ALS rammer motoriske nerveceller, har vi kunnet bruke det samme apparatet til å simulere hva som skjer med de kortikale glutamaterge nervecellene under frontotemporal demens (FTD), som kan ledsage den motoriske delen av ALS-sykdommen. Denne fleksibiliteten er en stor fordel. Her kan vi så ut kortikale glutamaterge nerveceller i to adskilte grupper slik at vi kan undersøke kontaktene mellom dem under nøye kontrollert betingelser, og vi kan også se på hvordan gliacellene virker inn på dem. Vi kjenner ikke til noen andre in vitro sykdomsmodeller der hele ALS-FTD sykdomsspekteret kan studeres på den måten vi kan her.
5) Vi har også jobbet med et helt annet prosjekt, som tar sikte på å utvikle en metode for fremtidig behandling av ALS. Fordi ALS rammer motoriske nerveceller som er spredt langs hele lengden av hjernestammen og ryggmargen, er det vanskelig å se for seg et inngrep som kan rettes spesifikt mot denne celletypen. Men en måte å få dette til på er å levere noe til skjelettmuskelcellene som de motoriske nervecellene har kontakt med. Som nevnt under punkt 2 bruker motoriske nervecellene denne kontakten ikke bare til å aktivere muskelcellene, men også til å forskyne seg med livsviktige vekst- og overlevelsesfaktorer fra muskelcellene. Vi kan derved bruke denne forskyningsmekanismen til å levere andre ting til de motoriske nervecellene. For eksempel, dersom vi injisere et molekylært «fraktsystem» inne i en muskel, vil dette kunne medføre transport av f.eks. medikamenter fra muskelen til de motoriske nervecellene.
I vårt prosjekt har vi brukt et stoff som vi har jobbet med i mange år – dextranmolekyler – som vi vet transporteres fra muskelcellene til de motoriske nervecellene via nervefibrene. Vi har nå laget en spesiell kjemisk forbindelse mellom dextranmolekylene og molekyler som kan hindre at et mutert gen uttrykkes. Forbindelsen har vi gjort lys-følsomt, slik at effekten kan skrues på med infrarødt lys etter at dextranmolekylene har fraktet «medisinen» frem til de motoriske nervecellene. Vårt håp er at denne metoden kan brukes i fremtiden til å «skru av» muterte gener hos ALS pasienter, og derved potensielt stoppe sykdomsutviklingen.
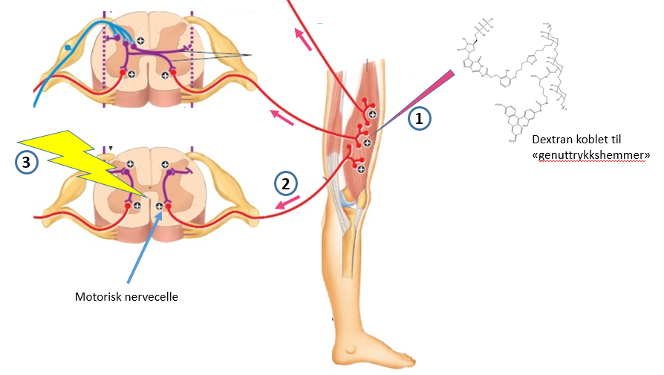
Figur 2. Prinsippet bak vår behandlingsmodell. Dextranmolekyler med tilkoblet «genmutasjonshemmende»-molekyler injiseres i en muskel (1). Disse fraktes til de motoriske nervecellene i ryggmargen (2). Belysning av ryggmargen med svakt infrarødt lys (3) frigjør molekylene inne i de motoriske nervecellene og hindrer at det muterte genet uttrykkes.
6) Som en siste aktivitet har vi jobbet videre med våre planer om å etablere en nasjonal biobank av iPS-celler som kan brukes til behandling av mange i den norske befolkningen. iPS-cellene i denne biobanken vil ha den egenskapen at de kan «matches» til pasienter på samme måte som man «matcher» benmarg til ulike pasienter. Når biobanken er etablert, vil vi ha en nasjonal resurs som kan brukes til å lage alle kroppens celletyper, for eksempel motoriske nerveceller, ut i fra den enkelte pasientens behov for celler. Dersom man finner en måte å erstatte motoriske nerveceller på hos ALS-pasienter i fremtiden, vil vi da stå klare til å kunne lage dem fra iPS-cellene i biobanken.
Samtlige av disse prosjektene er blitt støttet av ALS Norge. Denne støtten er uhyre viktig for at vi kan holde disse prosjektene gående, og gjør oss i stand til å gjøre nye fremskritt samt samarbeide med utenlandske ALS forskere mot å finne måter å behandle og en dag helbrede ALS på.
Vi takker ALS Norge for støtten og gjør oss klar til fornyet innsats på vegne av alle som er berørt av ALS.
Oslo, 08. januar 2021 Joel C. Glover